Истинная скорость - реакция
Cтраница 2
 |
Зависимость концентрации ным, так и отрицательным в зави-веществ А и В от времени симости от того, каким методом. [16] |
Если относить изменение концентрации вещества к бесконечно малому промежутку времени, то можно определить истинную скорость реакции v в данный момент времени. [17]
Зволинский и Эйринг [1334], рассматривая на простой модели процесс мономолекулярного распада, нашли, что истинная скорость реакции приблизительно на 20 % меньше того значения, которое получается в предположении равновесного распределения. [18]
Когда скорость реакции намного превосходит скорость подвода реагентов, макроскопическая кинетика определяется процессами транспорта и не отражает истинной скорости реакции на поверхности, ее зависимости от температуры, концентрации и других параметров. [19]
Если реакция протекает во внутренней диффузионной области ( в случае пористого катализатора), то Wr будет отличаться от истинной скорости реакции. [20]
Анализируя (6.9.4), заключаем, что если fea: 3a, то макроскопическая скорость реакции определяется в основном значением величины ka и мало отличается от истинной скорости реакции. [21]
Представляет также интерес и второй вопрос: каким образом можно из ряда значений концентраций и отвечающих им моментов времени, которые дает опыт, вычислить истинную скорость реакции. На эти вопросы нам даст ответ основной закон химической кинетики, который иногда называют также законом действующих масс. Содержание этого закона мы выясним, рассмотрев с молеку-лярно-кинетической точки зрения реакции между газообразными веществами. [22]
Резонансные эффекты проявляются в сопряженных и ароматических системах и по существу представляют собой отклонение скоростей реакций, вычисленных в приближении локализованных связей и зарядов, от истинных скоростей реакций. Пространственный фактор существен в тех случаях, когда либо в исходных веществах, либо в переходном состоянии имеются пространственные напряжения. Наконец, роль растворителя в органических реакциях очень велика, но обычно реакционные серии рассматривают для какого-либо одного или для родственных растворителей и тем самым сводят на нет действие этого фактора. [23]
Так как скорость реакции непрерывно изменяется ( как правило, уменьшается), то, вычисляя изменение концентрации A [ S ] за какой-то конечный промежуток времени, мы не можем определить истинную скорость реакции. [24]
В диффузионной области скорость реакции, так же как коэффициент диффузии, слабо зависит от температуры, а в кинетической скорость реакции изменяется по закону Аррениуса w w0 C-ERT При протекании гетерогенных каталитических реакций в контактных аппаратах скорость процессов определяется истинной скоростью реакции на поверхности катализатора и скоростью диффузии реагирующих веществ к контакту. [25]
Электродные процессы являются гетерогенными, и их кинетика зависит от электрических переменных, характеризующих условия на поверхности. Истинная скорость реакции пропорциональна току, и в качестве электрической переменной всегда выбирается потенциал электрода. Поэтому зависимость ток - потенциал характеризует кинетические закономерности. Кинетика электродных процессов изучалась с начала столетия, концепция перенапряжения была введена в 1899 году. Тафель предложил использовать кривые перенапряжение - логарифм плотности тока еще в 1905 году, но современные представления появились несколько позже. Они были впервые изложены Батлером в 1924 году. [26]
Анализ полученных этим автором результатов показал, что каталитическое влияние пикриновой кислоты в начальных стадиях разложения жидкости мало. Поэтому истинная скорость реакции в данном случае может быть выведена из начальной скорости. [27]
При увеличении истинной скорости реакции глубина проникновения уменьшается; согласно ( II, 70), она обратно пропорциональна корню квадратному из скорости реакции. Макроскопическая же скорость реакции пропорциональна произведению из истинной скорости реакции на глубину проникновения и, следовательно, увеличивается пропорционально корню квадратному из истинной скорости реакции. [28]
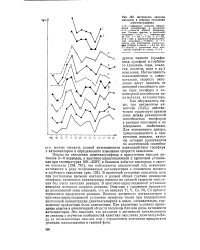 |
Активность окислов-металлов в реакции окисления диметилсульфяда. [29] |
В проточной установке окисление шло при постоянном времени контакта и разной общей глубине конверсии тиоэфира, активность катализатора оценена по средней скорости окисления без учета кинетики реакции. В проточно-циркуляционной установке измерена истинная скорость реакции. Опытами с удалением продуктов из реакционной зоны показано, что на окислах Ti, V, Co, Ni, Си процесс тормозится продуктами реакции. Поэтому активность катализаторов в проточно-циркуляционной установке измерена также при поддержании постоянной концентрации диметилсульфида в цикле, следовательно, торможение продуктами было одинаковым. При различных условиях проведения опытов в кинетической области получены близкие ряды активности катализаторов. Это означает, что различия в активности катализаторов не связаны с неучетом особенностей кинетики окисления диметилсульфида на исследованных окислах или с отравлением контактов продуктами реакции, находящимися в газовой фазе. [30]
Страницы: 1 2 3 4