Возбужденное электронное состояние - молекула
Cтраница 3
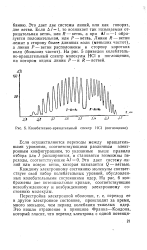 |
Колебательно-вращательный спектр НС1 ( поглощение. [31] |
Каждому электронному состоянию молекулы соответствует свой набор колебательных уровней, обусловленных колебательными состояниями ядер. На рис. 6 изображены две потенциальные кривые, соответствующие невозбужденному и возбужденному электронному состоянию молекулы. [32]
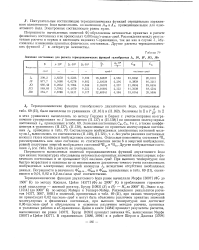 |
Значения постоянных для расчета термодинамических функций газообразных J2, JO, JF, JC1, JBr. [33] |
Погрешности вычисленных значений термодинамических функций двухатомного йода при низких температурах обусловлены неточностью принятых значений молекулярных и физических постоянных и не превышают 0 01 кал / моль - град. При высоких температурах они быстро возрастают в основном из-за невозможности достаточно точного учета составляющих возбужденных электронных состояний молекулы J2 вследствие отсутствия необходимых данных. Фзооо и Фадоо, приведенных в табл. 69 ( II), оцениваются в 0 01, 0 02 и 0 2 кал / моль - град соответственно. [34]
Однако Барроу [652, 645, 646] и Михель [2871] показали, что эти полосы обусловлены переходами между возбужденными электронными состояниями молекулы ОН. [35]
На основании этих исследований Малликен пришел к выводу, что основным электронным состоянием молекул галогенов является состояние 1Eg, а нижними электронными состояниями, которые коррелируют с состояниями 2Р 2Р атомов галогенов, должны быть состояния 3Пи, ЧТы, 1Пе и 3ПЙ [2987], и показал, что три последних состояния должны быть состояниями отталкивательного типа, причем переходам 1П - Х11 соответствуют континуумы, наблюдаемые в спектрах поглощения всех галогенов в ближнем ультрафиолете или видимой области. На основании теоретических соображений и анализа экспериментальных данных Малликен показал ( см. [2987]), что в возбужденных электронных состояниях молекул галогенов характер спин-орбитальной связи соответствует случаю Гунда с или близок к нему. [36]
Таким образом, из электронно-колебательно-вращательных спектров можно определить колебательные и вращательные постоянные не только основного, но и возбужденных электронных состояний. Это дает возможность вычислить энергию молекулы на различных колебательных и вращательных уровнях как для основных, так и для возбужденных электронных состояний молекулы. [37]
Однако в связи с тем, что только небольшая часть возможных электронных состояний молекул является стабильной, определение верхнего предела в статистической сумме по электронным состояниям в общем виде практически невозможно. Тем не менее это не приводит к существенным затруднениям и ошибкам в расчетах термодинамических функций двухатомных газов, поскольку большая часть возбужденных электронных состояний молекул имеет высокие энергии и вклад этих состояний становится существенным лишь при таких температурах, когда молекулы газа практически полностью диссоциируют на атомы. Кроме того, в высоких электронных состояниях потенциальные кривые молекул имеют слабо выраженный минимум. В таких электронных состояниях молекула имеет небольшое число колебательных и вращательных уровней энергии, а статистические суммы по колебательно-вращательным состояниям малы по сравнению с величиной QKOJt. [38]
Метод встречных пучков нашел широкое применение в лазерной спектроскопии высокого разрешения. Он используется для исследования тонкой и сверхтонкой структур атомных спектров, для измерения изотопических сдвигов уровней, для наблюдения эффекта Штарка и эффекта Зеемана, для спектроскопии возбужденных электронных состояний молекул, в ряде сложных экспериментов, посвященных исследованию динамики переходных процессов, например распаду возбужденных состояний. [39]
Можно надеяться, что книга окажется полезной химикам ( и, возможно, другим специалистам), которые занимаются изучением механизмов химических реакций. В книге используется метод молекулярных орбиталей ( МО), поскольку, вероятно, в настоящее время это наиболее мощный метод для трактовки молекулярных структур, химической связи и возбужденных электронных состояний молекул. При этом предполагается, что читатель имеет представления о теории МО, теории групп и спектроскопии. [40]
Термодинамические функции газообразных соединений водорода и его изотопов с галогенами, вычисленные в интервале температур 293 15 - 6000 К без учета межмолекулярного взаимодействия, приведены в табл. 44 - 46, 53 - 55, 62 - 64 и 71 - - 73 II тома Справочника. Термодинамические функции были вычислены для природной смеси изотопов хлора и брома по усредненным значениям молекулярных постоянных. Возбужденные электронные состояния молекул газов в расчетах не учитывались, поскольку при температурах, не превосходящих 6000 К, вклад соответствующих составляющих ничтожно мал. [41]
Что касается возбужденных электронных состояний молекул, то за редкими исключениями они не учитывались в этих расчетах. [42]
Приближенные оценки энергий возбуждения и постоянных SiO в этих состояниях, данные авторами работы [2535], не надежны и нуждаются в проверке и уточнении. Ввиду этого, а также вследствие высоких энергий возбуждения, эти состояния SiO в настоящем Справочнике не рассматриваются. Еще четыре возбужденных электронных состояния молекулы SiO с энергиями возбуждения, превосходящими 52 500 еж 1, были обнаружены Барроу и Роулинсоном [3539, 660,662] при исследовании спектра поглощения окиси кремния в области вакуумного ультрафиолета. Эти состояния также не рассматриваются в настоящем Справочнике. [43]
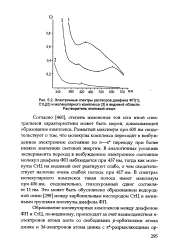 |
Электронные спектры растворов диафена ФП ( 1, СтЦ ( 2 и молекулярного комплекса ( 3 в видимой области. [44] |
Согласно [460], степень изменения той или иной спектральной характеристики может быть мерой, доказывающей образование комплекса. Размытый максимум при 600 нм свидетельствует о том, что молекулы комплекса переходят в возбужденное электронное состояние по п-я переходу при более низких значениях световой энергии. В аналогичных условиях эксперимента переход в возбужденное электронное состояние молекул диафена ФП наблюдается при 437 нм, тоща как молекулы СтЦ на видимый свет реагируют слабо, о чем свидетельствует наличие очень слабой полосы при 417 нм. В спектрах молекулярного комплекса такая полоса имеет максимум при 406 нм, следовательно, гипсохромный сдвиг составляет 11 нм. Это может быть обусловлено образованием водородной связи [298] между карбонильным кислородом СтЦ и амин-ными группами молекулы диафена ФП. [45]
Страницы: 1 2 3 4