Макроскопическая стадия
Cтраница 1
Макроскопические стадии наблюдаются и в жидкофазном окислении углеводородов, катализированном солями металлов переменной валентности. Впервые было замечено [46, 99], что при окислении керосина в присутствии нафтена марганца катализатор находится в растворенном состоянии и инициирует реакцию только в начале окисления. Затем катализатор полностью выпадает в осадок и не принимает участия в окислении. Таким образом, катализированное окисление углеводородов часто протекает в две стадии: в первой стадии катализатор находится в растворе и, инициируя цепи, ускоряет окисление; во второй - он выпадает в осадок и не принимает участия в инициировании цепей. Однако такое резкое разделение реакции на две стадии встречается не всегда. [1]
Макроскопическими стадиями называются последовательные стадии, разграниченные во времени. Последующая стадия практически не идет, пока не окончится предыдущая. При этом каждая из таких стадий может представлять собой сложную многостадийную реакцию. [2]
Наличие макроскопических стадий в катализированном окислении углеводородов обусловлено, по-видимому, изменением физико-химических свойств катализатора в углеводородном растворе по мере накопления молекулярных продуктов окисления. В уксуснокислых растворах, где катализатор в течение исего процесса полностью диссоциирован на ионы, макроскопической стадийности не наблюдается. В углеводородном растворе катализатор весьма чувствителен к полярным веществам. Его растворимость в значительной степени зависит от полярности среды, аниона, входящего в состав соли. Образование комплексов с продуктами окисления может влиять на активность катализатора. [3]
Наличие макроскопических стадий в катализированном окислении углеводородов обусловлено, по-видимому, изменением физико-химических свойств катализатора в углеводородном растворе по мере накопления молекулярных продуктов окисления. В уксуснокислых растворах, где катализатор в течение всего процесса полностью диссоциирован на ионы, макроскопической стадийности не наблюдается. В углеводородном растворе катализатор весьма чувствителен к полярным веществам. Его растворимость в значительной степени зависит от полярности среды, аниона, входящего в состав соли. Образование комплексов с продуктами окисления может влиять на активность катализатора. Из продуктов окисления, оказывающих влияние на агрегатное состояние катализатора, важная роль принадлежит кислотам. На опыте неоднократно наблюдалось, что в реакциях окисления катализатор выпадает в осадок в виде солей кислот, образующихся при окислении. Однако выпадение катализатора в осадок в ходе реакции окисления - результат не простой обменной реакции между солью и кислотой, а сложный процесс, в котором наряду с кислотами принимают участие и другие продукты окисления, в частности вода. Макроскопические стадии в процессе катализированного окисления ( изменение валентного состояния катиона, выпадение катализатора в осадок) обычно протекают очень быстро. Добавки некоторых соединений могут значительно удлинять стадии валентных превращений. Так, марганцовый катализатор в реакции окисления синтина в начале тормозит окисление ( Мп2 обрывает цепи), затем, превратившись в Мп34, ускоряет процесс окисления, но довольно скоро выпадает в осадок. При окислении синтина в присутствии смесей нафтенатов марганца и калия стадии валентных превращений марганца удлиняются: период торможения увеличивается с 10 до 40 мин. [4]
В первой макроскопической стадии окисления металл катализатора переходит в высшее валентное состояние. Об этом можно непосредственно судить по изменению окраски деканового раствора, которая становится характерной для трехвалентных производных марганца и кобальта. Образовавшееся из катализатора промежуточное соединение дает в углеводороде устойчивые при комнатной температуре растворы. В ходе реакции интенсивность окраски раствора увеличивается и проходит через максимум. Одновременно в системе начинается выпадение осадка соли металла в исходном двухвалентном состоянии. [5]
Являются ли макроскопические стадии в гомогенном катализе спецификой катализа бромистым водородом или это общий механизм катализа. Майзус и автор [ 8 исследовали с помощью метода разогрева реагирующей смеси окисление пропана, катализированное окислами азота. Добавка небольших количеств окислов азота позволяет значительно снизить температуру проведения реакции, что способствует сохранению ценных кислородсодержащих продуктов окисления - перекисей, альдегидов, кеюнов, спиртов. [6]
Наконец третья макроскопическая стадия составляет лишь окисление ацетальдегида двуокисью азота до конечных продуктов, включающих метшшитрит, нитрометан и метилнитрат, воду, окись азота, двуокись углерода и формальдегид. [7]
 |
Изменение отношения разогрева IT к скорости w реакции Н2 - ( - С12 в присутствии добавок азота ( / и кислорода ( 2 при 310 С ( по данным А. М. Мар-кевича. [8] |
Следовательно, первая макроскопическая стадия является гомогенной. [9]
После изучения макроскопических стадий была составлена система параллельных и последовательных реакций в возможном механизме процесса. [10]
Представление о макроскопических стадиях и особой, определяющей, роли начального периода позволяет нам высказать новую точку зрения по вопросу о природе предельных выходов продуктов в некоторых цепных реакциях. В самом деле, почему так распространены факты прекращения образования тех или иных продуктов реакции задолго до того, как израсходованы исходные вещества: Эти предельные выходы но могут быть объяснены термодинамическими соотношениями, так как обратные реакции в рассматриваемых условиях, как правило, практически не идут. Весьма часто нельзя объяснить эти предельные выходы конкуренцией двух процессов - процесса образования некоторого продукта и процесса его расходования. Так, например, обстояло дело с предельными выходами ацетона при окислении пропана в присутствии НВг или с выходами уксусной кислоты при окислении этапа в присутствии того же катализатора. Естественно, что ацетон и уксусная кислота в условиях рассматриваемых опытов ни в каких последующих превращениях практически но участвуют. [11]
Гипотеза б макроскопических стадиях распространяется и на положительный катализ. Среди катализаторов также найдутся такие, которые оказывают влияние лишь на первую макроскопическую стадию, а после образования достаточного количества разветвляющих агентов катализаторы уже будут неэффективны. [12]
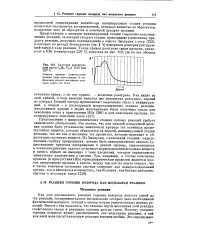 |
Разогрев реагирующей смеси С3Нв, 02 и НВг при 220 С. [13] |
Представление о макроскопических стадиях цепных реакций требует химического обоснования. Это значит, что для каждой конкретной реакции должна быть установлена химическая природа тех активных промежуточных веществ, которые образуются на первой, инициирующей стадии реакции, так же как и механизм тех процессов, которые приводят к образованию активных веществ. По Эмануэлю, с начальной стадией, определяющей глубину превращения, должно быть непосредственно связано существование пределов превращения в данной системе, выражающихся в остановке реакции задолго до полного израсходования исходных веществ и ничего общего не имеющих с теми пределами, которые определяются химическим равновесием в системе. [14]
Представление о макроскопических стадиях цепных реакций требует еще химического обоснования. Это значит, что для каждой конкретной реакции должна быть установлена химическая природа тех активных промежуточных веществ, которые образуются па первой, инициирующей стадии реакции, так же как и характер и механизм тех процессов, которые приводят к образованию активных веществ. Однако уже сейчас плодотворность представления о макроскопических стадиях совершенно очевидна. В частности, по мысли Н. М. Эмануэля, с наличием начальной стадии, определяющей глубину превращения ( см. выше), должно быть непосредственно связано существование пределов превращения в данной системе, выражающихся в прекращении ( остановке) реакции задолго до полного израсходования исходных веществ и ничего общего не имеющих с теми пределами, которые полагаются химическим равновесием в системе. Другим примером может служить упоминавшаяся нами ранее ( стр. Этот и подобные факты, очевидно, нужно приписать тем химическим изменениям в системе ( в случае гомогенно-каталитических реакций определяющимся процессами, происходящими в инициирующей стадии), в результате которых она переходит в новое состояние, в котором могут осуществляться лишь процессы, отвечающие новой, последующей стадии, или прекращается всякое химическое превращение. Заметим, что тривиальный случай такого превращения реакции задолго до достижения термодинамического равновесия мы имеем в раз-ветвленно-цепных реакциях горения различных газов, когда после выгорания определенного количества исходного горючего система приходит к такому состоянию, в котором процессы обрыва цепей становятся преобладающими над процессами разветвления и реакция прекращается ( затухает), несмотря на наличие в системе и горючего, и кислорода в количествах, значительно превышающих их равновесные концентрации. По-видимому, нужно считать, что предельные явления, установленные для реакций медленного окисления и выражающиеся в полном прекращении реакции при определенной глубине превращения или в переходе от одной макроскопической стадии реакции к другой, также определяются соотношениями скоростей отдельных элементарных процессов, достигающими некоторых предельных ( критических) значений на определенных стадиях химического превращения в системе. [15]
Страницы: 1 2 3 4