Влияние - алкильная группа
Cтраница 2
Оказалось, что влияние электронодонорных алкильных групп проявляется значительно меньше в ЦТМ, чем в бензоле. Так, введение одной метильной группы в ЦТМ увеличивает скорость изотопного обмена водорода в 9 раз, а этильной группы в 5 раз, в то время как одна метильная группа в бензольном кольце увеличивает скорость водородного обмена в 150 раз. Три этильные группы в циклопентадиенильном кольце ЦТМ активируют дейтерообмен меньше, чем одна метильная группа в бензольном кольце. Константа скорости водородного обмена триэтилЦТМ возрастает в 126 раз, а скорость водородного обмена 1 3 5-триметилбензола так велика, что измерить ее в тех же условиях не удается. [16]
Такая же последовательность влияния алкильных групп наблюдается по значениям констант диссоциации соответствующих бензойных кислот. [17]
Каким образом можно объяснить влияние алкильных групп на устойчивость этих алкенов. [18]
Ниман и сотрудники рассмотрели влияние алкильных групп на накожную и остаточную эффективность ДДТ двух серий соединений - N-н-алкил - п-хлорбензол - и М - н-алкил-д-бромбен - зол-сульфо - п - хлоранилидов. [19]
 |
Влияние природы алкильных групп и растворителей при прото - и галоиддеметаллированни ртутно - и олозоорганических соединений. [20] |
Гилен и Насельский103 сравнили влияние разных алкильных групп в реакциях протолиза и галоидирования ртутноорганиче-ских и оловоорганических соединений ( табл. 31), а также в различных реакциях ртутноорганических соединений, обсуждаемых в других разделах этой книги. [21]
При помощи неэмпирического расчета влияния алкильных групп на основность и кислотность установлено [34], что поляризуемая алкильная группа может делокализовывать как отрицательный, так и положительный заряд. Эффективность дело-кализации увеличивается с ростом размера радикала: Me Et i - Pr t - Bu. Рассчитанные энергии переноса протона согласуются с данными по кислотности и основности в газе. [22]
При помощи неэмпирического расчета влияния алкильных групп на основность и кислотность установлено [34], что поляризуемая алкильная группа может делокализовывать как отрицательный, так и положительный заряд. Эффективность дело-кализации увеличивается с ростом размера радикала: Me Et [ i - Pr t - Bu. Рассчитанные энергии переноса протона согласуются с данными по кислотности и основности в газе. [23]
Если бы в этом случае влияние алкильных групп заключалось только в индукционной поляризации молекул, они бы расположились по своему действию в обратной последовательности, - как это показано выше. Отсюда Бекер и Натан сделали вывод, что кроме индуктивного эффекта алкильные группы могут проявлять еще один вид влияния. По предположению Бекер а и Натана этот эффект обусловливается способностью а-электронов С - Н - связей участвовать во взаимодействии с я-электронами соседней кратной связи или ароматического ядра. [24]
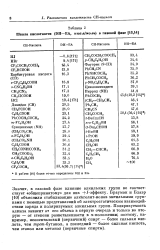 |
Шкала кислотности ( DH-ЕА, ккал / моль в газовой фазе. [25] |
Значит, в газовой фазе влияние алкильных групп не соответствует общепринятому для них / - эффекту. Брауман и Блаэр [10] объяснили стабилизацию алк оголят-ионов метильными группами с помощью представлений об электростатическом взаимодействии заряда и поляризуемых алкильных групп. [26]
 |
Кривые поглощения ( Люис и Кальвин. [27] |
Люис и Кальвин9 объясняют это влияние алкильных групп индуктивным эффектом, возмущающим электронные пары соединения без существенного изменения их характера. Влияние ауксохромных групп, не содержащих свободных электронных пар, находит более точное объяснение в теории о-п-сопряжения. [28]
Бантон и Верной [108] изучили влияние электронодонорной алкильной группы на примере одного из имеющих важное биологическое значение фосфатов Сахаров. На кривой скорость - рН для гидролиза a - D-глюкопиранозил-фосфата в воде наблюдается подъем от очень низких скоростей в щелочном растворе до плато при рН около 5, при котором концентрация моноаниона достигает максимального значения; однако затем вместо нового спада, который должен был бы наблюдаться, если бы вследствие увеличения кислотности среды функция гидролизующейся частицы перешла бы к неионизованной молекуле моноэфира, вновь наблюдается повышение скорости, которое в области рН от 4 до 0 5, когда происходит постепенное увеличение доли реакции с участием неионизованной молекулы, осуществляется по линейному закону. Поэтому в данном случае реакция неионизованной молекулы с водой ( или в воде) вместо того, чтобы быть более медленной, чем реакция аниона ( что наблюдается, например, при гидролизе монометил-фосфата), на самом деле происходит с гораздо большей скоростью. Наряду с изменением природы субстрата от моноаниона до пеионизованной молекулы происходит и изменение типа расщепляемой связи. Согласно данным изотопного метода с использованием 180, в моноанионе разрывается связь фосфор - кислород, а в неионизованной молекуле - связь углерод - кислород. Несмотря на то что молекулярность этих реакций по воде не установлена, очевидно, можно полагать, что гидролиз и моноаниона a - D-глюкопиранозил-фосфата, и моноаниона метилфосфата имеет одинаковый механизм ВАС 1 - Одинаково также и положение расщепляющейся связи ( в обоих случаях фосфор - кислород), а скорости имеют один и тот же порядок величины. [29]
Подробный количественный анализ показывает, что влияния алкильных групп в положениях, аналогичных пара-положениям в бензоле, могут быть настолько велики, что их нельзя объяснить индукто-электромерными взаимодействиями, так что зуше-ственную роль может играть сверхсопряжение; но этот вывод нельзя считать вполне надежным. [30]
Страницы: 1 2 3 4