Изменение - энергия - сольватация
Cтраница 3
И тем не менее все эти выводы, несмотря на свою кажущуюся убедительность, ошибочны. Дело заключается в том, что изменение работы выхода электрона при переходе от одного металла к другому или изменение энергии сольватации при переходе от одного растворителя к другому компенсируется соответствующим изменением гальвани-потенциала. [31]
 |
Значения Е0 Д2Аох, Igy0, lgy0OCH -, lgT03JI - и А2СОл. для НС1 при 25. [32] |
Проведенные нами расчеты показывают, что для растворов НС1 не только величина lg TO, но и lg ТЭ0Л не являются линейной функцией I / O. Линейность соблюдается лишь до 80 % неводного растворителя, что объясняется тем, что в смесях, содержащих менее 80 % неводного растворителя, сольватные оболочки ионов состоят только из молекул воды, и изменение энергии сольватации определяется изменением лишь диэлектрических свойств и состава. В растворах с большим содержанием неводного растворителя происходит пересольватация, образование смешанных сольватных оболочек, что и приводит к нарушению линейной зависимости. [33]
Следовательно, она включает в себя и химический потенциал электрона в металле, и стандартный химический потенциал иона водорода. Поэтому, когда выносят W0 в константу, не зависящую якобы от скачка потенциала, то допускают ошибку, ибо скачок потенциала непосредственно определяется химическими потенциалами электрона и иона водорода, в частности энергией сольватации протона. Как известно, изменение энергии сольватации протона должно вызывать соответствующее изменение энергии активации. Прирост энергии активации равен половине прироста энергии сольватации протона, и этим обычно объясняется зависимость перенапряжения от природы растворителя. Однако при этом не учитывают, что изменение энергии сольватации влечет за собой изменение скачка потенциала, и эти два влияния строго равны и противоположны. Другими словами, можно сказать, что от энергии сольватации зависит, если пользоваться предложенной М. И. Темкиным терминологией, идеальная энергия активации и не зависит реальная энергия активации, которая определяет величину перенапряжения. [34]
Закономерность, наблюдаемая при сочетании ионных эффектов среды, может быть использована для косвенного суждения об относительной энергии сольватации ионов в исследуемых растворителях. А - т практически принимает одно и то же значение для всех сильных кислот. Этот факт наводит на мысль, что свободная энергия переноса отвечает, в основном, изменению энергии сольватации протона. Дальнейшее уточнение основывается на вычислении химической энергии сольватации протона в воде и неводных растворителях [6], а также на вычислении тун из константы равновесия перехода протона от Н30 к SH в комбинации с членом, который учитывает различие электростатического окружения в двух средах [7] [ подобно рассуждениям Борна, уравнение ( VII. Эти два приближения приводят к хорошо согласующимся результатам. [35]
 |
Потенциалы нулевого заряда поверхности разных металлов в водородной шкале. [36] |
Исследования строения двойного слоя в неводных растворах проведены в основном на ртутном электроде в спиртах, амидах и некоторых апротонных растворителях, таких, как диметилсульф-оксид, пропиленкарбонат и ацетонитрил. Основные закономерности теории строения двойного слоя соблюдаются и в этом случае. Специфическая адсорбция анионов в сильной степени зависит от природы апротонного растворителя, что связано с изменением энергии сольватации в них анионов по сравнению1 с водой. Абсолютные значения дифференциальной емкости в апротонных растворителях обычно ниже, чем в воде, и не коррелируют с диэлектрической проницаемостью растворителя. [37]
 |
Константы скорости реакции СН1 Х - СН3Х Г в СН3ОН и ДМФ. [38] |
Из табл. 2.36 очевидно, что порядок изменения нуклеофильности малых анионов почти полностью обращается при переходе из протонных в диполярные апротонные растворители. Ионная ассоциация может скрывать эту перестановку ( гл. HS ( 1 - Вг - С1 - F -) отражает тогда инверсию либо степени делокализации заряда, либо ряда изменения энергии сольватации. [39]
Вторым типом дифференцирующего действия является изменение относительной силы кислот различных природных групп под влиянием любых растворителей, особенно не содержащих гидроксильных групп. При переходе к таким дифференцирующим растворителям сила кислот одной природной группы изменяется примерно одинаково, но отлично от изменения силы кислот другой природной группы. Такое действие является следствием того, что величины 70ионов и бТомолекул близки для кислот одной природы и сильно отличаются для кислот различной природы. Изменение активности ионов кислот разной природы объясняется изменением энергии сольватации анионов. Изменение коэффициентов активности недиссоциированных молекул объясняется различием в энергии присоединения к ним молекул растворителей, которое становится особенно значительным при переходе от растворителя одной природы крастворителю другой природы. [40]
Совокупность корреляций структуры и реакционной способности, которая составляет основное содержание физической органической химии, основана прежде всего на исследованиях органических реакций в растворе. Многие правила и корреляции существенно определяются энергией молекул реагентов, электронной стабилизации или дестабилизации активированного комплекса, промежуточных продуктов и конечных продуктов реакции. В гетеролитических реакциях растворитель играет зачастую важную роль, оказывая влияние на стабилизацию ионов, ионных пар и промежуточных ионизированных частиц вследствие сольватации. Поэтому иногда оказывается затруднительным дифференцировать вклад в изменение реакционной способности при введении некоторого заместителя, обусловленный собственным стабилизирующим влиянием заместителя на активированный комплекс, и действующее в благоприятном направлении изменение энергии сольватации. В таких случаях приходится прибегать к теоретическим или полуэмпирическим расчетам, которые не всегда просты и не всегда достаточно надежны. Ясно, что было бы в высшей степени полезно знать энергии органических ионов по данным экспериментов в газовой фазе. Весьма благоприятной оказалась бы и возможность осуществления в газовой фазе хотя бы некоторых реакций, обычно протекающих в растворе. [41]
Следовательно, она включает в себя и химический потенциал электрона в металле, и стандартный химический потенциал иона водорода. Поэтому, когда выносят W0 в константу, не зависящую якобы от скачка потенциала, то допускают ошибку, ибо скачок потенциала непосредственно определяется химическими потенциалами электрона и иона водорода, в частности энергией сольватации протона. Как известно, изменение энергии сольватации протона должно вызывать соответствующее изменение энергии активации. Прирост энергии активации равен половине прироста энергии сольватации протона, и этим обычно объясняется зависимость перенапряжения от природы растворителя. Однако при этом не учитывают, что изменение энергии сольватации влечет за собой изменение скачка потенциала, и эти два влияния строго равны и противоположны. Другими словами, можно сказать, что от энергии сольватации зависит, если пользоваться предложенной М. И. Темкиным терминологией, идеальная энергия активации и не зависит реальная энергия активации, которая определяет величину перенапряжения. [42]
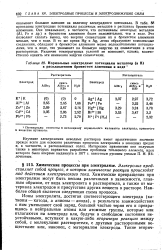 |
Нормальные электродные потенциалы металлов ( в В в расплавленном бромистом алюминии и воде. [43] |
В табл. 88 приведены электродные потенциалы различных металлов в расплавах бромистого алюминия. Они показывают, в частности, что потенциалы некоторых металлов ( Al, Fe и др.) в бромистом алюминии более положительны, чем в воде. Это позволяет сделать вывод, что энергии сольватации их в бромистом алюминии меньше, чем в воде. В то же время Hg и Ag, легко образующие комплексные соединения, обладают в бромистом алюминии менее положительными потенциалами, чем в воде, что указывает на большую энергию их сольватации в этом растворителе. Различие температурных условий не позволяет, однако, количественно оценить эти изменения энергии сольватации. [44]
Прежде чем приступить к изложению и обсуждению дальнейших результатов, целесообразно обратить внимание на отличие эффектов растворителя в электрохимической кинетике от соответствующих эффектов в кинетике гомогенных химических реакций. Эти изменения могут быть довольно большими, и они маскируют при этом более тонкие эффекты, определяемые изменением энергии реорганизации. В электрохимических же реакциях положение существенно иное. При равновесном потенциале свободная энергия исходного состояния - например, сольватированный ион водорода электрон в металле, и конечного состояния ( газообразный Н2) равны, и это равенство сохраняется при замене одного растворителя на другой. Иными словами, изменение энергии сольватации при переходе от одного растворителя к другому автоматически компенсируется вызванным этим изменением сдвигом равновесного скачка потенциала металл - раствор, так что уровень энергии исходного состояния в целом не меняется. Таким образом, сравнение кинетики в разных растворителях при равновесном потенциале - или, в более общем случае, при одинаковом перенапряжении - есть сравнение в изознергетических условиях. [45]
Страницы: 1 2 3 4