Учет - конфигурационное взаимодействие
Cтраница 3
Термин конфигурационное взаимодействие ( часто сокращаемый до KB) ведет свое начало именно от термина электронная конфигурация, поскольку переход к учету конфигурационного взаимодействия означает введение линейной комбинации конфигурационных функций состояния, соответствующих различным электронным конфигурациям. Получаемая при этом функция уже не отвечает какой-либо одной конфигурации. В связи с тем, что слово взаимодействие здесь не несет прямого физического смысла, не отображает какое-либо физическое взаимодействие частиц, оно, естественно, в данном употреблении не вполне удачно. [31]
Теория СТС ароматических свободных радикалов была разработана многими авторами, использовавшими как методы локализованных пар, так и метод молекулярных орбит с учетом конфигурационного взаимодействия. [32]
Чтобы оценить влияние точности выбранной волновой функции на результат, был произведен аналогичный расчет с использованием волновой функции Хиджи-каты [10], полученной с учетом конфигурационного взаимодействия. [33]
Поскольку по теории Хюккеля в нулевом приближении спиновые плотности на ядрах щелочных металлов в ионных парах также малы, можно ожидать, что расчеты с учетом конфигурационных взаимодействий приведут к отрицательным спиновым плотностям на этих ядрах. [34]
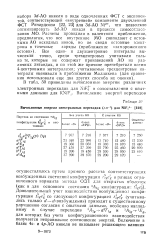 |
Вычисленные энергии спектральных переходов ( смг1 для NiF64 . [35] |
Дополнительный учет взаимодействия & возбужденнол конфигурации tige & со следующей конфигурацией 1 кеЛе ( учитывались только d - af - возбуждения) приводит к существенному улучшению согласия с опытными данными, особенно наглядному в случае переходов 3A2g - - a3T g и 3A2g - 1ES, для которых без учета конфигурационного взаимодействия получается неправильное соотношение энергий. [36]
Таким образом, в рамках основного электронного состояния таких радикалов не удается найти объяснения для протонного расщепления. Однако учет конфигурационного взаимодействия [20] приводит не только к качественному объяснению такого явления, но и к количественному согласию с опытом. [37]
Метод PCILO [139-141] отличают две особенности. Во-первых, задача учета конфигурационного взаимодействия решается в рамках метода теории возмущений Релея - Шредингера. Во-вторых, в качестве молекулярных орбиталей используются орбитали, локализованные иа связях. Причем в качестве локализованных орбиталей выбираются не линейные комбинации орбиталей ССП МО ЛКАО, а произвольные связывающие и аптисвязывающие орбитали, строящиеся, например, из гибридизоваиньтх атомных орбиталей. [38]
ППДП, благодаря чему им удалось интерпретировать электронные спектры некоторых углеводородов и их гетероаналогов. В этих расчетах применялся также ограниченный учет конфигурационного взаимодействия, который будет подробно рассмотрен в разделе, посвященном расчетам я-электронных систем. [39]
Это очень незначительное понижение объясняется слабостью конфигурационного взаимодействия. С другой стороны, только благодаря учету конфигурационного взаимодействия удается получить очень хорошее согласие с экспериментом при сделанном выборе параметров. Появляются систематические отклонения, поскольку теория предсказывает положения а-полосы в области более длинных волн. Однако изменив соответствующим образом параметры, удается получить также для а - и 5-полос ароматических углеводородов хорошее согласие с экспериментом, как и для jo - полос. Обозначим посредством i - /) конфигурации, в которых г-я молекулярная орбиталь заменяется на некоторую до того не занятую / - ю орбиталь, причем молекулярные орбитали, занятые в основном состоянии системы, нумеруются в порядке убывания энергии, а незанятые молекулярные орбитали - в порядке возрастания энергии. Когда в линейной комбинации конфигураций доминируют две конфигурации 1 - - 2) и 2 - 1, то говорят, что такие состояния имеют а - или р-харак-тер в зависимости от того, мала или велика интенсивность соответствующего электронного перехода. В ароматических углеводородах р -, а - или ( 3-характер соответствующих полос весьма четко определен. [40]
Это очень незначительное понижение объясняется слабостью конфигурационного взаимодействия. С другой стороны, только благодаря учету конфигурационного взаимодействия удается получить очень хорошее согласие с экспериментом при сделанном выборе параметров. Появляются систематические отклонения, поскольку теория предсказывает положения а-полосы в области более длинных волн. Однако изменив соответствующим образом параметры, удается получить также для ос - и Р - ПОЛОС ароматических углеводородов хорошее согласие с экспериментом, как и для / ьполос. Обозначим посредством i - / конфигурации, в которых z - я молекулярная орбиталь заменяется на некоторую до того не занятую; - ю орбиталь, причем молекулярные орбитали, занятые в основном состоянии системы, нумеруются в порядке убывания энергии, а незанятые молекулярные орбитали - в порядке возрастания энергии. Когда в линейной комбинации конфигураций доминируют две конфигурации 1 - 2 и 1 2 - 1, то говорят, что такие состояния имеют а - или р-харак-тер в зависимости от того, мала или велика интенсивность соответствующего электронного перехода. В ароматических углеводородах р -, а - или р-характер соответствующих полос весьма четко определен. [41]
Ван Брокховен [54, 130 - 132] экспериментально, а Соммердийк [125, 132 - 134] теоретически исследовали влияние проти-воионов на свойства дианионов трифенилена, трифенилбензола, декациклена и трифенил-с лш-триазина. При расчетах термов методом самосогласованного поля с учетом конфигурационного взаимодействия (3.1.3) использовали молекулярные орбитали хюккелевского типа, построенные, как описано в разд. Появление термически возбужденного триплетного состояния ди-аниона 2 4 6-трифенил-сылш - триазина в метилтетрагидрофуране, как предполагают, определяется конформацией, в которой катионы локализованы вблизи атомов азота. Расположение катиона над центром дианиона нельзя принять, поскольку для этой конформации расчет дает основное триплетное состояние, что противоречит экспериментальным данным. Расположение катионов над внешними кольцами также можно исключить, поскольку такие конфигурации приводят к термически недостижимым три-плетным состояниям, что также не согласуется с опытом. [42]
 |
Порядки я-связей в антрахиноноксадиазоле ХСП, антрахинонтиади-азрле ХСШ и антрахинонселенадиазоле XCIV, рассчитанные методом МО ЛКАО в приближении Хюккеля. [43] |
Расчет методом ОСП-МО ЛКАО Паризера - Парра - Полпла с учетом конфигурационных взаимодействий, выполненный для антрахиноноксадиазо-ла, указывает на еще большую, чем дает упрощенный метод МО ЛКАО, неравномерность распределения электронной плотности ( порядки связей: С-4-С-5 0 813; С-5-С-5а 0 482; С-5 а С-11 а 0 741; С-11 а - С-1. Длины связей в аитрахиноноксадиазо-ле, вычисленные из порядков, рассчитанных методом ССП-МО ЛКАО, лучше согласуются с найденными экспериментально. По-видимому, для антрахинонтиадиазола и антрахинонселенадиазола истинная степень нарушения выровненности связей в крайнем кольце также выше, чем дает расчет методом МО ЛКАО. [44]
 |
Порядки я-связей в антрахиноноксадиазоле ХСП, антрахинонтиади. [45] |
Страницы: 1 2 3 4