Фенилкетонурия
Cтраница 1

Фенилкетонурия является одним из наиболее распространенных заболеваний обмена фенилаланина и связана с отсутствием в организме фермента фенилаланин-4 - гидроксилазы, которая катализирует превращение фенилаланина в тирозин. В организме при этом скапливается большое количество фенилаланина, вызывающее ряд вторичных биохимических реакций. Так, взаимодействие фенилаланина с пируватом приводит к образованию фенилпировиноградной кислоты, которая в больших количествах выводится с мочой. [2]
Фенилкетонурия развивается как результат потери способности организма синтезировать фенилаланингидроксилазу, катализирующую превращение фе-нилаланина в тирозин. Развитие болезни можно предотвратить, если значительно снизить или исключить прием фенилаланина с пищей с самого рождения ребенка. [3]
Фенилкетонурия может служить примером довольно часто встречающейся генетической болезни, которую легко распознать и которая поддается лечению. [4]
Фенилкетонурия ( фенилпировиноградная олигофрения) развивается как результат потери способности организма синтезировать фенилаланин-4 - монооксигеназу, катализирующую превращение фенилаланина в тирозин. Решающим доказательством метаболического блока при фенил-кетонурии являются данные о накоплении фенилаланина в тканях. [5]
Фенилкетонурия наблюдается при отсутствии нормальной фонилаланин-гидроксилазной активности. В этих условиях фенилаланин не может превращаться в тирозин ( см. фиг. Это очень серьезное нарушение обмена; оно приводит к резкому замедлению умственного развития детей. [6]
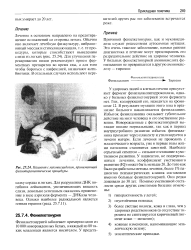 |
Пациент смуковисцидозом, принимающий физиотерапевтические процедуры. [7] |
Фенилкетонурией заболевает примерно один из 10 000 новорожденных белых, а каждый из 80 таких младенцев является носителем. [8]
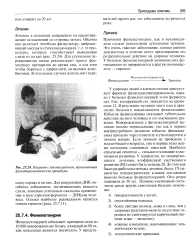 |
Пациент смуковисцидозом, принимающий физиотерапевтические процедуры. [9] |
Причиной фенилкетонурии, как и муковисци-доза служит рецессивная аутосомная мутация. Это очень тяжелое заболевание, однако ранняя диагностика и лечение могут предотвратить его разрушительное действие на здоровье человека. [10]
При врожденной фенилкетонурии наблюдается недостаточное количество фермента, который превращает путем гидроксилирования фе-нилаланин в тирозин. [11]
 |
Вариации возраста начала заболевания хореей Гентингтона. [12] |
Почему больным фенилкетонурией повезло, что фенилаланин является незаменимой аминокислотой. [13]
Как доказать, что фенилкетонурия контролируется рецессивным геном. [14]
Как доказать, что фенилкетонурия не сцеплена с полом. [15]
Страницы: 1 2 3 4