Флуоресценция - соединение
Cтраница 2
Определение алюминия основано на измерении интенсивности флуоресценции соединения алюминия с салицилаль-о-аминофенолом [1, 2] при рН 6 2 - 6 4 без отделения больших количеств кадмия. Определению мешают железо в количестве, большем 1 мкг, и медь в количестве, большем 5 мкг, в 10 мл раствора. [16]
В табл. 41 приведены спектральные характеристики поглощения и флуоресценции соединений 5 красителей, применяемых в рекомендуемых далее методах, и перечень соответствующих разрешающих устройств типовых приборов, выпускаемых в Советском Союзе. При измерениях экстрактов, не содержащих ацетона на спектрофотометре СФ-4 значение А макс соединения уточняют опытным путем. [17]
Однако приводимые в их работах сведения относятся к флуоресценции соединений, адсорбированных на целлюлозе. [18]
Поглощенная доза в этом случае прямо пропорциональна уменьшению интенсивности флуоресценции соединения, возбуждаемой ультрафиолетовым светом после облучения. В этом случае мерой радиационных нарушений в сцинтилляторе служит изменение числа вспышек ( сцинтилляций) под действием ионизирующей радиации. [19]
Целесообразно описать стадии, необходимые для определения квантового выхода флуоресценции соединения с помощью относительного метода. [20]
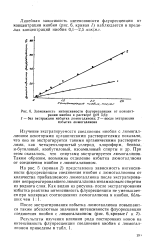 |
Зависимость интенсивности флуоресценции от концентрации ниобия в растворе ( рН 5 6. [21] |
На рис. 5 ( кривая 2 представлена зависимость интенсивности флуоресценции соединения ниобия с люмогаллионом от количества прибавляемого люмогаллиона после экстрагирования непрореагировавшего люмогаллиона изоамиловым спиртом из водного слоя. [22]
Ввиду изложенного мы сочли необходимым, несмотря на скудность данных о флуоресценции соединений жирного ряда, тем не менее эти данные подытожить, дополнить собственными наблюдениями и систематизировать в таблицы; наша задача - внести возможную ясность в вопрос о характере флуоресценции рассматриваемого класса веществ. При составлении таблиц мы старались относиться к имеющимся данным критически и приводить только те из них, которые получены в результате наблюдения ( насколько можно судить) химически чистых препаратов. Данные наших таблиц относятся к свечению только в видимой части спектра - флуоресценция соединений жирного ряда в коротковолновой части спектра, насколько нам известно, не изучена, если не считать приведенных в таблице опытов Штарка, давших отрицательный результат. При описании флуоресценции приходится, как ясно из вышесказанного, довольствоваться указанием воспринимаемого глазом цвета свечения или констатацией отсутствия видимой флуоресценции. [23]
Как видно из табл. 1, наиболее сильное мешающее влияние на флуоресценцию соединения ниобия с люмогаллионом оказывают титан и железо. В ходе определения необходимо отделение мешающих элементов. [24]
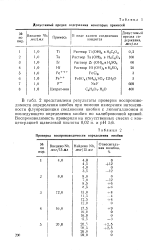 |
Допустимый предел содержания некоторых примесей. [25] |
В табл. 2 представлены результаты проверки воспроизводимости определения ниобия при помощи измерения интенсивности флуоресценции соединения ниобия с люмогаллионом и последующего определения ниобия по калибровочной кривой. [26]
Авторы работы [11] указывают, что наряду с преимущественным влиянием водородной связи на флуоресценцию соединений типа XVII немаловажное значение имеют и другие факторы, такие, как диполь дипольное, диполь-поляризационное и дисперсионное взаимодействия. [27]
Гаврилова и Красновский [107] установили, что полярные молекулы не влияют на спектры поглощения и флуоресценции соединений, не содержащих магния ( феофитина и фталоцианина), и поэтому приписывают отмеченный выше эффект связыванию этих молекул через остаточные валентности магния. [28]
Хотя в настоящее время еще имеется мало данных о природе флуоресценции, создается впечатление, что флуоресценция соединения в ряде случаев зависит от его фазового состояния и степени дисперсности. [29]
Стократный избыток Са, Al, Ре ( П) и Fe ( I) лишь незначительно снижает интенсивность флуоресценции внутрикомпдексного соединения магния с хюмомагнезоном. Меньшие количества указанных катионов не мевают определению магния. При тысячекратном избытке мешают кроме того Li, Ва, Мо, Те, Zr, V, As, Pd. При стократном избытке Са чувствительность реакции становится 0 1 мкгМд, но внесение винной кислоты в количестве 0 01 % дает возможность определятъМд в присутствии тысячекратного избытка Са с чувствительностью реакции 0 01 икгМд в 5 мл. [30]
Страницы: 1 2 3 4