Хроматографирова-ние
Cтраница 1
Хроматографирова-ние проводят в эксикаторе на хромато-графической бумаге, вырезанной в виде лопаток ( см. рис. 22) следующих размеров: а б10 см, в45 см, г 15 см. Отмеренные микропипеткой 0 005 мл или 0 01 мл пробы, наносят на узкую часть лопатки на расстоянии 1 см от широкой части, как описано на с. Пятно высушивают на воздухе и бумажную лопатку устанавливают в эксикаторе, погружая ее узкий конец на 1 - 2 см в чашку Петри, в которую налит растворитель. [1]
Хроматографирова-ние на анионитах различных марок в оксалатной, тартратной или цитратной формах позволяет [183] разделять ионы третьей и второй групп катионов. [2]
Разработаны методы хроматографирова-ния свободных металлов при сверхвысоких тысячеградусных температурах. [3]
Действительно, применив последовательное хроматографирова-ние на силикагеле и окиси алюминия, Рождественская, Кроль, Жердева и Кучерявая [62, 63] выделили из трансформаторных масел фенольной и фурфурольной очистки узкие фракции, содержащие 90 - 100 % сернистых соединений, главным образом сульфидов. Авторы предположили на основании таких характеристик, как показатель преломления, удельный вес, молекулярный вес и удельная дисперсия, что эти сульфиды состоят из би - и трицикланов, являющихся, по-видимому, производными тиофана. [4]
Лучше всего провести предварительное хроматографирова-ние, которое подскажет, надо ли пробу концентрировать или разбавлять, не слишком ли много в ней межклеточного вещества, какие типы примесей в ней имеются, какова полярность анализируемых веществ и какая система растворителей предпочтительна. [5]
Поскольку в процессе жидко-жидкостного хроматографирова-ния какие-то несмешивающиеся жидкости до некоторой степени растворяются друг в друге, то для продления жизни хроматогра-фической колонки необходимы специальные меры. Подвижную фазу необходимо предварительно насыщать неподвижной, чтобы избежать удаления последней из колонки в процессе хроматогра-фирования. Полное равновесие двух несмешивающихся жидкостей в действительности достигается труднее, чем это можно предположить. Ввиду ограниченной растворимости двух фаз контакт между ними при смешивании относительно плохой. Описанный ниже метод, как было установлено, позволяет достичь состояния удовлетворительного равновесия двух фаз. Заключается он в следующем. Обе фазы ( обезгаженные или насыщенные азотом) помещаются под азотом в закрытый контейнер в отношении от 10 / 1 до 50 / 1 ( подвижная / неподвижная), быстро смешиваются магнитной мешалкой до образования мутной воронки и перемешиваются несколько часов или ночь. После этого фазы разделяются; подвижная фаза используется для хроматографирования, а неподвижная - возвращается в цикл. [6]
Конец реакции устанавливают хроматографирова-нием в тонком слое окиси алюминия, растворитель бензол - хлороформ 2: 3, проявитель концентрированная серная кислота. [7]
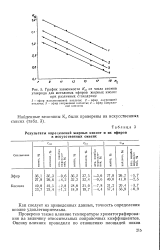 |
График зависимости K. s от числа атомов углерода для метиловых эфиров жирных кислот. [8] |
Проверено также влияние температуры хроматографирова-ния на величину относительных поправочных коэффициентов. [9]
Во избежание разрушения 3 4-бензпирена хроматографирова-ние проводят в затемненном помещении, а колонку защищают цилиндром из черной бумаги с вертикальной прорезью. По мере прохождения раствора доливают небольшими порциями бензол. Вытекающие из колонки фракции собирают в зависимости от цвета и интенсивности флуоресценции. Растворы фракций переводят в колбы Вюрца и отгоняют бензол. Остаток на дне колбы растворяют в небольших порциях октана до полного прекращения флуоресценции вновь вливаемого растворителя, доводя общий объем до 4 - 5 мл. [10]
Возможно разделение лантана и актиния хроматографирова-нием на бумаге растворов этих элементов в водном бутиловом спирте, ацетилацетоне и уксусной кислоте. [11]
Концентрация углеводородов в элюате за время хроматографирова-ния обычно не остается постоянной. При микрохроматографировании минеральных масел первые порции элюата содержат относительно много углеводородов. Поэтому при отборе в первые 3 - 4 микроприемника испарение двух-трех капель элюата дает 0 2 - 0 7 мг углеводородов. Затем концентрация углеводородов в фильтрате постепенно падает, и соответственно количество петролейного эфира, которое необходимо испарить, чтобы набрать 0 5 - 1 0 мг микрофракции, возрастает. При десорбции последних порций метано-нафтеновых углеводородов накапливание 0 2 - 0 3 мг микрофракции занимает более 15 мин. Затем наступает период, когда поступление углеводородов с элюатом практически прекращается на довольно большой промежуток времени - 20 - 50 мин. Это явление перерыва в поступлении углеводородов хорошо заметно и четко фиксируется при регистрации времени смены микроприемников. После некоторого промежутка времени углеводороды опять начинают поступать с элюэнтом, но гораздо медленнее, чем в первый период. После перерыва отбирают еще 2 - 3 микрофракции по 0 2 - 0 4 мг, на этом десорбцию метано-нафтеновых ( и самых первых ароматических) фракций заканчивают. [12]
По имеющимся данным [78] в процессе хроматографирова-ния в координационной сфере металлов протекают конкурирующие реакции между силанолами или силоксанами и лигандами. [13]
Реакция проходит неполностью, продукты выделялись хроматографирова-нием на силикагеле. Основным продуктом ( 33 %) оказался метиловый эфир 96, образующийся, очевидно, при внедрении метоксид-иона из растворителя в первично образующийся эпоксид. [14]
Продукты силанизации стабильны, и перед хроматографирова-нием обычно их не выделяют из реакционной смеси. Силани-зующие же реагенты настолько летучи, что даже при большом их избытке они появляются в самом начале хроматограммы и не мешают определению разделяемых компонентов. [15]
Страницы: 1 2 3 4