Льюисовский центр
Cтраница 2
 |
Теплоты адсорбции СО иг поликристэллических переходных металлах. [16] |
Образование гидроксильного покрова оксидов приводит к исчезновению льюисовских центров. Гидроксильные группы, образующиеся на поверхности, способны проявлять кислотно-основные свойства в бренстедовском смысле. [17]
 |
Колебательные полосы поглощения в ИК-спектрах пиридина ( П и 2 6-диметилпиридина ( ДМП. адсорбированных в виде ионов на бренстедовских и. лъюисовских ( Л кислотных центрах. [18] |
Было показано [30, 31], что в присутствии льюисовских центров не происходит предпочтительной адсорбции пиридина на бренстедовских центрах. Это явление оказывается чрезвычайно полезным в тех случаях, когда требуется селективно отравить бренстедовские центры в присутствии льюисовских. Хотя метилзамещенные пиридины обладают большей основностью, чем пиридин, их реакционная способность в отношении льюисовских центров ниже; это объясняется стерическими затруднениями, обусловленными экранированием реакционноспособного центра ( атом N) метальными группами. [19]
По мнению Туркевича и Оно, роль льюисовских центров не ограничивается лишь увеличением кислотности соседних ОН-групп и эти центры оказывают более непосредственное влияние на каталитические превращения. Трудно сказать, насколько точно эти авторы выяснили природу промежуточных соединений, но они показали, что надо говорить не об активных центрах вообще, а о центрах, на которых протекают конкретные реакции. Центры могут быть активны в одних превращениях и не участвовать в других. Например, кривая зависимости активности цеолитов NH4Y ( степень обмена 50 и 90 %) в реакции изомеризации циклопропана в пропилен при 70 С от Гакт имеет два максимума [87]: один при 300 - 400 С, другой около 650 С. Поэтому был сделан вывод, что существуют два механизма изомеризации с участием бренстедовских и дегидроксилированных центров. [20]
Для получения оптимального катализатора необходимы определенное соотношение между количеством льюисовских центров и числом слабокислых изолированных гидроксильных групп и определенная структура твердых тел. В наших экспериментах энстатит, очевидно, наиболее полно отвечал этим требованиям. [21]
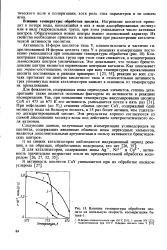 |
Влияние температуры обработки цеолита на начальную скорость изомеризации бу-тена-1. [22] |
Концентрация гидроксильных групп при этом тоже снижается, а концентрация льюисовских центров увеличивается. Это доказывает, что только бренсте-довские центры являются первичными центрами катализа. Как показывает цис: транс-соотношение, остающееся при всех температурах прокаливания цеолита постоянным и равным примерно 1 [23, 55], реакция во всех случаях протекает на центрах кислотного типа и относительная активность трех упомянутых выше катализаторов зависит в основном 6т температуры их прокаливания. [23]
Более того, Хиршлер [58] ставит под сомнение само существование льюисовских центров, указывая, что следы кислорода либо в реагентах, либо на поверхности катализатора легко могут окислить трифе-нилметан до трифенилкарбинола; бренстедовская кислотность должна привести к образованию карбоний-иона. Как известно, такая реакция действительно происходит в кислотных средах, в которых сила кислоты больше, чем в 50 % - ной H2SO4, и процесс этот ускоряется при действии света. Удаление гидрид-иона заменяется окислением, катализируемым кислотой, и для этого необходимы сильные бренстедовские кислотные центры. [24]
Однако даже небольшое содержание примесей может изменять эти свойства; например, льюисовские центры находят на пористом стекле викор [30], что может быть связано с присутствием примеси алюминия. Хотя высокая удельная поверхность силикагеля делает его ценным носителем, сам силикагель как катализатор весьма инертен. Он слабо активен в разложении спиртов [31], возможно из-за примеси ионов А13, и в большинстве случаев его значение как катализатора несущественно. Тем не менее гамма-облучение или радиоактивное облучение в ядерном реакторе придает ему некоторую каталитическую активность. Если двуокись кремния хорошо обезгажена, облучение создает также кислотные центры, катализирующие реакции изомеризации двойной связи и полимеризацию олефинов. [25]
В образцах, обработанных около 600 С, начинает уменьшаться и концентрация льюисовских центров. При этой же температуре начинается разрушение решетки цеолита. Число кислотных центров, найденное по результатам титрования пиридином образцов, прогретых при температуре выше 400 С, также составляет только 10 % концентрации центров, найденной по титрованию аммиаком. Такое различие связано не с большей основностью аммиака, а скорее всего с недоступностью каналов клиноптилолита для адсорбированных молекул пиридина. Потвидимому, молекулы пиридина взаимодействуют исключительно с теми гидроксильными группами, которые расположены на внешней поверхности этого цеолита. [26]
Для измерения концентрации гидроксильных групп ( бренстедов-ские центры) и трехкоординированного алюминия ( льюисовские центры) было использовано взаимодействие пиридина с цеолитами. [27]
Температура термообработки была достаточно высокой, поэтому можно было бы ожидать также присутствия льюисовских центров, однако данных о них в работе не приводится. [28]
Для алюмо - и никельсиликатов Озаки и Кимура [80] нашли, что изомеризацию ведут льюисовские центры, а донорами протонов являются молекулы олефина, хемосорбиро-ванные на этих центрах. Таким образом, в некоторых случаях льюисовские центры могут давать тот же эффект, что и бренстедовские. [29]
Эти факты показывают, что при введении воды в систему в бренстедов-ские центры превращаются только сильные льюисовские центры. Из использованных нами цеолитов, по-видимому, только LaX, СеХ и СаХ имеют достаточное количество лыоисовских центров, способных под действием воды превратиться в бренстедовские центры. [30]
Страницы: 1 2 3 4