Термодинамическая гибкость
Cтраница 2
Одним из средств, позволяющих оценить термодинамическую гибкость полимерной цепи, является исследование параметров, связанных с размерами клубка изолированной макромолекулы. [16]
Этот факт подчеркивает определяющее влияние на термодинамическую гибкость взаимодействий ближнего порядка. В полимерах винилового ряда сфера влияния полярных заместителей затрагивает вращение вокруг одинарных связей, что усиливает действие сил отталкивания и повышает Д Л Поэтому гибкость этих полимеров несколько снижается при введении полярных заместителей. [17]
Быстрота затухания ориентационных корреляций связана с термодинамической гибкостью полимерной цепи, отражает протяженность ближнего порядка в цепи. [18]
Хорошая совместимость полиокса с многими пластиками и высокая термодинамическая гибкость его цепей позволяют использовать его в качестве пластификатора, в частности для повышения прочности и эластичности желатиновых слоев в современных фото - и кинопленках. Будучи относительно новым материалом, полиокс далеко не исчерпал возможностей своего практического использования. [19]
Согласно поворотно-изомерной концепции, предложенной Волькен-штейном, термодинамическая гибкость класса гибкоцепных полимеров возникает за счет реализации набора разных поворотно-изомерных кон-формаций цепи. [20]
Исследование двойного лучепреломления в потоке позволяет определить термодинамическую гибкость нативной ДНК ( см. стр. Гибкость характеризуется персистентной длиной примерно прямолинейного участка. Длине 500 А отвечает примерно 150 мономерных звеньев. По-видимому, макромолекулу нативной ДНК нельзя считать зигзагообразной, состоящей из строго прямолинейных участков. Скорее она имеет червеобразную форму с непрерывно меняющейся кривизной. [21]
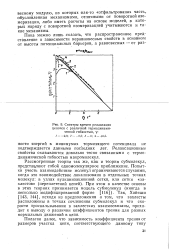 |
Спектры времен релаксации цепочек с различной термодинамической гибкостью, у. [22] |
Релаксационные свойства оказываются довольно тесно связанными с термодинамической гибкостью макромолекул. [23]
Поэтому для характеристики эластичности полимерных материалов необходимо знать термодинамическую гибкость макромолекул ( определяющую наибольшую возможную деформацию) н скорость развития деформации. [24]
Поэтому для характеристики эластичности полимерных материалов необходимо знать термодинамическую гибкость макромолекул ( определяющую наибольшую возможную деформацию) и скорость развития деформации. [25]
Изложенные выше понятия и определения, связанные с равновесной термодинамической гибкостью, относятся к полимерным цепям, кон-формационные свойства которых определяются только взаимодействиями ближнего порядка. В реальных цепях в растворе ( даже при бесконечном разбавлении) существуют объемные взаимодействия между удаленными по цепи, но случайно сблизившимися в пространстве звеньями одной и той же цепочки. [26]
Обусловлено это тем, что именно в случае эластомеров высокая термодинамическая гибкость изолированных макромолекул сочетается со сравнительно малым межмолекулярным взаимодействием в полимере. Количественным выражением этого взаимодействия является плотность энергии когезии - величина, в случае жидкости численно равная энергии, необходимой для испарения 1 см3 вещества. [27]
Поэтому для характеристика эластичности полимерных мате риалов необходимо знать термодинамическую гибкость макрсшо лекул ( определяющую наибольшую возможную деформацию) i скорость развития деформации. [28]
Поэтому для характеристики эластичности полимерных мат риалов необходимо знать термодинамическую гибкость макрос лекул ( определяющую наибольшую возможную деформацию) скорость развития деформации. [29]
Следует отметить, что природа заместителей оказывает незначительное влияние на термодинамическую гибкость. [30]
Страницы: 1 2 3 4