Анализируемая кислота
Cтраница 2
Навеску анализируемой кислоты разбавляют дистиллированной водой. [16]
Навеску анализируемой кислоты, взятую по объему с относительной точностью до 1 - 5 %, содержащую от 0 001 до 0 010л г платины, помещают в гонкую фторопластовую чашку, прибавляют 2 мл раствора сернокислого натрия и выпаривают па водяной бане, помещенной в бокс из оргстекла, почти досуха. Прибавляют 2 мл царской водки л выпаривают досуха. После разделения фаз экстракт фильтруют через небольшой слой гигроскопической ваты г, кювету с толщиной колориыетрируемого слоя 1 см и измеряют оптическую плотность на спектрофотометре СФ-4А относительно органической фазы контрольного опыта, проведенного через весь ход анализа. [17]
 |
Основные технические характеристики концентратомера КСО-3. [18] |
Расход анализируемой кислоты и олеума через датчик - 2 0 5 л / мин. Прибор отградуирован при температуре кислоты 56 С. [19]
Пробу анализируемой кислоты тщательно перемешивают и отбирают 45 - 50 г, помещая в предварительно взвешенную фарфоровую чашку. Кислоту упаривают на песочной бане до объема около 1 - 1 5 мл. [20]
Например, если анализируемая кислота X массой 1 225 г полностью нейтрализуется гид-роксидом натрия массой 1 000 г, то молярную массу ее эквивалента находят из пропорции: 1 225 / 1 000М ( / ЭКв ( Х) Х) Ш ( NaOH), откуда для кислоты М ( / экв ( Х) Х) 49 г / моль. [21]
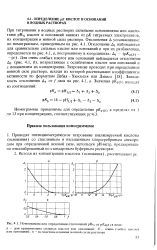 |
Номограмма для определения отклонений pHi / 2 от pKa ( i в воде. [22] |
Проводят потенциометрическое титрование анализируемой кислоты ( основания) со стеклянным и насыщенным хлорсеребряным электродом при определенной ионной силе, используя рН - метр, предварительно откалиброванный по стандартным буферным растворам. [23]
Помещают 10 мл анализируемой кислоты, отмеренные пипеткой, в мерную колбу емкостью 100 мл, доливают воду до метки и перемешивают. [24]
Помещают 10 мл анализируемой кислоты в кварцевую или фарфоровую чашку и упаривают на песочной бане до прекращения выделения паров серной кислоты. Осадок растворяют при нагревании в 3 мл соляной кислоты. Раствор нейтрализуют ам - миаком до полного выделения осадка гидроокиси железа и приливают еще 1 5 мл избытка раствора аммиака. [25]
Определив окислительно-восстановительный потенциал анализируемой кислоты и массовую долю оксидов азота по градуи-ровочным таблицам, находят массовую долю воды в анализируемой среде. [26]
Пиролиз тетраметиламмониевых солей анализируемых кислот проводится в испарителе при 330 - 365 С. Показано, что выход эфиров при пиролизе тетраметиламмониевых солей понижается при величинах пробы, меньших 0 05 мг. [27]
Растворяют 4 г анализируемой кислоты в воде и доводят объем раствора до 20 мл. Переносят его в электролизер, добавляют 0 2 мл раствора ртути, содержащего 1 мг Hg11 в 1 мл, и вытесняют кислород током инертного газа. [28]
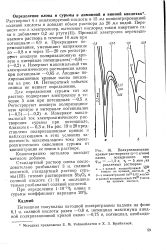 |
Поляризационные кривые растворения ( vl в / мин олова, осажденного при рэл - 1 2 в, Т ], 5 мин JHS различных растворов. [29] |
Растворяют 4 г анализируемой кислоты в 15 мл концентрированной соляной кислоты и доводят объем раствора до 20 мл водой. Проводят электролиз перемешиваемого раствора в течение 10 мин при потенциале - 0 9 в. Прекращают перемешивание, уменьшают напряжение до - 0 8 в и через 15 - 20 сек регистрируют анодную поляризационную кривую в интервале потенциалов ( - 0 8) - ( - 0 2) в. Измеряют максимальный ток электрохимического растворения олова при потенциале - 0 65 в. Пятикратный избыток свинца не мешает определению. [30]
Страницы: 1 2 3 4